Signes d'épilepsie rolandique chez les nourrissons. L'épilepsie rolandique chez l'enfant et ses symptômes
L'épilepsie rolandique fait référence aux formes bénignes de la maladie, caractéristiques de l'enfance. Selon les statistiques, sur 100 mille enfants de moins de 14 ans sur 21 enfants, au moins une fois dans leur vie, une crise d'épilepsie bénigne pourrait être observée. Parmi tous les patients, les psychothérapeutes et les psychiatres qui sont traités pour l'épilepsie, la forme rolandique est présente chez 15% d'entre eux.
Les premiers signes de la maladie apparaissent chez les enfants âgés de 2 à 3 ans (avant cet âge, la pathologie n’est pas caractéristique). Le pic des symptômes tombe entre 7 et 9 ans. À l'adolescence, la maladie passe progressivement. C'est pour cette raison qu'il fait référence à des formes bénignes: la guérison complète est possible. Les attaques cessent complètement à 15-18 ans. Les adultes ne souffrent pas de cette pathologie.
Quel épisindrome est considéré bénin?

Selon l’opinion courante des épileptologues, l’épilepsie bénigne en est une dont les symptômes ne nécessitent pas de traitement ou ne permettent pas une intervention minime et disparaissent sans conséquences.
Une saisie unique, qui survient souvent avec une forme rolandique de la maladie, permet certes de la classer comme une pathologie complète, mais il est de l’habitude chez les spécialistes de l’attribuer aux crises. La Ligue internationale antiépileptique a suggéré que l’épilepsie rolandique soit attribuée aux syndromes auto-limités. Il s'en va tout seul et ne cause pas de dommages structurels au cerveau. Lorsque le cerveau mûrit, son épileptogénicité diminue. Au début, il est courant de séparer les convulsions du nouveau-né (jusqu'à 18 mois); convulsions fébriles et syndromes focaux de bébés jusqu'à 2 ans; symptômes végétatifs chez les enfants de 3 à 4 ans; attaques focales de la pathologie rolandique de 3 à 15 ans (moins de 18 ans). Les crises bénignes ne sont jamais observées à l'adolescence et à l'âge adulte.
Facteurs qui augmentent le risque d'épilepsie rolandique
La maladie affecte la zone de Roland du cerveau. Le foyer de crise prend naissance dans les cellules nerveuses de la région temporale . Dès que les neurones du cerveau des enfants mûrissent, leur excitabilité diminue immédiatement. Par conséquent, la forme rolandique est généralement attribuée à des maladies plus jeunes, dont les symptômes disparaissent à mesure que l'enfant grandit.
À ce jour, aucune raison exacte n'a été établie qui provoque le développement de la pathologie.
Il existe une version selon laquelle l'un des facteurs peut être génétique: les enfants dont les parents souffrent d'une autre forme d'épilepsie augmentent le risque.
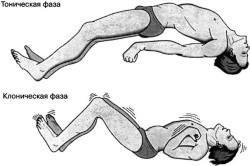
Types et symptômes d'attaques

L'épilepsie rolandique se caractérise par les types de crises suivants:
- partielle simple - détectée dans les contractions convulsives une à une face avant (touchant parfois le bras); engourdissement de la langue, des joues, du larynx;
- partiel partiel - accompagné de convulsions de muscles individuels, augmentation de la salivation, altération de la parole. Capable d'entrer dans une crise convulsive classique généralisée.
Pour l'épilepsie bénigne, les traits suivants sont inhérents:
- l'apparition d'attaques pendant le sommeil;
- présence d'aura devant les convulsions (fourmillements, picotements du larynx, de la langue, des gencives);
- une bande-son du genre "Gorge de gorge";
- la durée maximale de l'attaque est de trois minutes (la durée minimale pouvant être de plusieurs secondes, elle n'est donc pas toujours remarquée);
- déclin progressif des symptômes: les enfants plus âgés souffrent des manifestations de cette forme de pathologie beaucoup moins fréquemment que les enfants.
L'épilepsie rolandique chez les enfants est plus difficile que chez les écoliers. C'est lié à l'immaturité système nerveux enfance. Pendant la période de croissance active et de restructuration du corps, les attaques peuvent être répétées plusieurs fois par semaine ou même tous les jours.
Comment diagnostique-t-on l'épilepsie bénigne?

Comme les symptômes de la maladie ne sont pas exprimés de manière aiguë et surviennent dans 80% des cas pendant la nuit, le diagnostic est réalisé par électroencéphalographie. Vous pouvez ainsi suivre l'activité du cerveau des enfants et identifier les violations. Il est conseillé de mener une étude la nuit (la pathologie se manifestant rarement à l’heure de la journée). Polysomnographie assignée en plus.
Sur la base de quels signes un neurologue diagnostique-t-il?
- Ondes et pics de forte amplitude dans la zone temporale centrale;
- activité cérébrale normale fonctionnelle;
- des pics pointus alternent avec des vagues mourantes.
Les trois caractéristiques ci-dessus constituent un «complexe rolandique» complet.
Comment distinguer la forme pathologique de la pathologie des autres maladies?
L'épilepsie rolandique chez les enfants présente des symptômes similaires avec d'autres troubles neurologiques. Le plus souvent, il est confondu avec d'autres formes d'épilepsie résultant de lésions cérébrales, d'entsifalopatii, de méningite, de la présence de tumeurs. La marque de la pathologie: préservation de l'intelligence; activité cérébrale normale sur l'EEG; manque de trouble de la conduite. Si un spécialiste a des doutes quant au diagnostic, il peut vous prescrire une imagerie par résonance magnétique supplémentaire du cerveau (IRM).
Comment ne pas manquer les premiers symptômes de la maladie?

L'épilepsie bénigne est différente de l'adulte. Les premiers signes de pathologie peuvent être difficiles à déterminer chez jeune âge. Après tout, l’activité accrue des nourrissons est attribuée aux particularités de caractère et à certains troubles neurologiques. La personne a été d'avis que, pendant l'épilepsie, il y aura toujours de fortes crises. Mais ce n'est pas vrai: l'évolution de la maladie dépend de la forme et des caractéristiques individuelles de l'organisme. Les épileptologues savent que les patients sans crises se produisent dans la pratique. Et le terme «épilepsie» cache plus de soixante anomalies dans une clinique similaire.
Ce à quoi les parents devraient prêter attention (après tout, l'épilepsie bénigne rolandique peut être accompagnée de diverses crises généralisées ou signalée par les précurseurs bien avant l'exacerbation):
- Avec une forme incontrôlée, le bébé se fige et son regard devient "absent". Un picotement des paupières ou une légère inclinaison de la tête sont caractéristiques. L'attaque dure quelques secondes, après quoi l'enfant reprend ses activités normales. Souvent, les parents n'attachent pas d'importance à ces symptômes, ils écrivent tout comme une distraction habituelle. De telles attaques sont plus courantes chez les filles.
- Dans l'épilepsie convulsive, une crise classique est présente: tension musculaire, convulsions.
- Au cours d'une crise atonique, un enfant peut tout simplement perdre conscience. En plus de l'évanouissement habituel, il n'y a plus aucun symptôme indiquant l'épilepsie. Mais la maladie se développe. Par conséquent, si les parents ont remarqué que le bébé commençait à perdre régulièrement conscience, il faudrait immédiatement le montrer à un spécialiste.
- Spasmes musculaires - pendant le sommeil ou immédiatement après le réveil, l'enfant redresse involontairement ses jambes pendant quelques secondes, incline son torse ou sa tête vers l'avant, resserre le bras de la poitrine. Souvent, les manifestations cliniques ne se produisent que sur les muscles du cou. Puis le bébé commence à secouer la tête. Ces spasmes sont caractéristiques des jeunes enfants et, à l'âge de 5 ans, ils décèdent sans laisser de trace ou entrent dans une certaine forme d'épilepsie.
Les autres signes avant-coureurs comprennent:
- plaintes à mal de têtequi est accompagné de nausée;
- petit trouble de la parole - un enfant est incapable de dire quoi que ce soit pendant quelques secondes ou produit des sons déformés;
- perturbation du sommeil, surtout si le bébé se plaint de rêves effrayants.
Enfin, l'expert aidera à comprendre le diagnostic après la recherche.
Epilepsie bénigne rolandique en statistique

Au fil des années, dans l’étude des troubles pathologiques chez les jeunes enfants, des spécialistes ont découvert une certaine tendance dans le développement et les symptômes d’un type bénin de maladie:
- l'âge minimum du patient est de plusieurs mois;
- fréquence moyenne de fixation de la pathologie: 20 enfants sur 100 000;
- âge critique pour la manifestation de symptômes aigus - 7-9 ans;
- prévalence sexuelle - chez les garçons, la maladie est diagnostiquée 50% plus souvent que chez les filles;
- la cause principale du développement est un facteur génétique;
- gravité de l'évolution - chez 20% des patients, des épisodes simples ou doubles sont enregistrés toute leur vie; 20% - fréquent; 1% des patients présentent un risque supplémentaire de développer des syndromes sévères;
- l'état mental n'est pas altéré;
- l'état neurologique est normal;
- la fréquence des attaques nocturnes - 80% de tous les cas;
- des manifestations de -30% observent des symptômes hémifaciaux (raccourcissement de la lèvre inférieure, augmentation de la salivation, retard de la parole), les autres présentent des signes oro-laryngés (engourdissements de la joue, de la langue, du larynx; déformation de la parole);
- la sécurité de la conscience pendant les attaques - sans perturbation dans 2/3 des patients;
- la durée des attaques - jusqu'à 2 minutes dans la plupart des cas. Un tiers des patients peuvent être prolongés.
Avant qu'un enfant atteigne l'âge de seize ans, la pathologie bénigne se résorbe généralement. L'épilepsie bénigne peut durer jusqu'à 18 ans seulement chez 2% des patients.

Devrais-je traiter l'épilepsie bénigne? Les médecins n'ont pas de consensus. En effet, même en l'absence totale d'intervention, les symptômes cliniques des formes bénignes passent par eux-mêmes. Par conséquent, certains médecins estiment qu'il n'est pas nécessaire de prescrire des médicaments antiépileptiques.
Leurs adversaires insistent sur autre chose: les signes de la maladie, bien que non clairement définis, peuvent conduire à un pseudolennox (épilepsie focale atypique) s'ils ne sont pas traités. Parfois, il existe un autre diagnostic sous la forme rolandique. Et un traitement tardif peut aggraver l'état du patient.
Si le médecin décide traitement médicamenteux, on prescrit alors à l'enfant un seul médicament d'acide valproïque. Avec ses inefficacités individuelles, ils sont remplacés par le lévétiracétam. La carbamazépine est indiquée pour les enfants de plus de 7 ans.
Les enfants de moins de 3 ans nécessitent une attention particulière. Ils ont observé l'immaturité des cellules nerveuses, ce qui conduit à des attaques fréquentes. En cas de suspicion de pathologie, un neurologue procède à un examen.
Chaque parent doit connaître les méthodes de premiers secours dans l'attaque:
- pendant convulsions toniques ou des convulsions musculaires soulèvent l'enfant;
- pour éviter que la langue ne tombe, mettez l'enfant sur le côté et mettez quelque chose de doux dans la bouche;
- les crises prolongées nécessitent d'appeler salle d'urgence.

Pour éviter que les crises d'épilepsie ne se reproduisent, les médecins conseillent aux parents de se conformer aux méthodes de prévention. L'accent est mis sur la routine quotidienne. Les perturbations du sommeil entraînent une irritabilité excessive du système nerveux et provoquent des convulsions. L'enfant est obligé de dormir suffisamment.
Si le bébé a eu au moins un épisode d’attaque nocturne, il est conseillé de dormir dans la chambre d’un adulte plutôt que de le laisser seul dans la crèche. Pour dormir séparément, utilisez un moniteur pour bébé. Avec l'épilepsie rolandique, les crises épileptiques surviennent le plus souvent la nuit. Il est donc important de garder l'enfant sous contrôle pour éviter toute chute de la langue. La maladie a souvent une aura préliminaire (sons caractéristiques de la gorge). Par conséquent, étant proches, les parents seront en mesure de réagir à temps à une attaque.
Il est strictement interdit de violer le régime du sommeil des enfants (changer l'heure de la ponte, refuser à un âge précoce le repos). Le système nerveux des enfants devrait être capable de se détendre et de se reposer complètement. Il n'est pas nécessaire de regarder la télévision tout en jouant un enfant en jouant à des jeux informatiques. Il n'est pas recommandé de nourrir le bébé avant de se coucher.
Dans les cas difficiles, lorsque la prévention et le traitement médicamenteux n’ont pas eu d’effet positif, un spécialiste peut prescrire un régime cétogène. Il s'agit d'un régime alimentaire spécial, défini par le médecin en fonction de l'âge, du poids et de l'activité physique de l'enfant. Un schéma nutritionnel est en cours d'élaboration avec une prédominance de graisse et un contrôle des glucides et des protéines. Les cinq premiers jours du régime alimentaire du bébé devraient être à la clinique sous la supervision de médecins qui surveilleront le niveau de corps cétoniques dans le corps. Ils peuvent être produits en excès et sont des toxines. Ce n’est qu’après avoir étudié comment le corps de l’enfant réagit à un changement de nutrition, puis il est renvoyé à la maison et les parents reçoivent des recommandations pour une plus grande conformité alimentaire. Il est interdit de prendre une décision sur la nomination d'un régime cétogène. Cela se fait uniquement sous la direction d'un médecin!

Le mode de vie correct de la mère pendant la grossesse est important pour la prévention de l'épilepsie chez les enfants. Pendant cette période, les mauvaises habitudes (tabac, alcool) devraient être complètement exclues; Il est interdit de participer à des activités pouvant avoir un effet toxique sur le fœtus. Une bonne alimentation du futur bébé repose sur une alimentation saine, une activité physique rationnelle et un régime quotidien adapté.
Prévisions
Selon les statistiques recueillies, plus de 90% des enfants de plus de 13 ans ne présentaient pas de symptômes d'épilepsie bénigne rolandique. Seuls 2% des adolescents de 16 ans présentent des symptômes bénins.
L'étude a révélé que les facteurs indésirables pouvant influer sur la gravité et la durée de la pathologie sont l'apparition précoce (jusqu'à 4 ans) et plus tard (après 10 ans) de l'épilepsie.
Des attaques répétées se produisent souvent au cours des 4 premières années. Une longue période de rémission ne signifie pas une guérison complète: chez certains enfants, après plusieurs années d'absence, les crises peuvent réapparaître. À l'âge adulte, une seule manifestation de symptômes sous une forme bénigne est possible. Cela est souvent dû à des changements hormonaux dans le corps. Par conséquent, il est plus souvent observé chez les femmes alors que, dans l’enfance, la forme rolandique de la maladie est plus caractéristique du sexe masculin.
Selon les concepts modernes, l'épilepsie chez les enfants est un groupe de pathologies chroniques chroniques disparates du cerveau.
En règle générale, ils apparaissent:
- crises d'épilepsie spécifiques sous la forme de convulsions non provoquées qui se produisent sans raison sur le fond d'une santé complète;
- autres signes spécifiques ("petites convulsions") se manifestant par des troubles mentaux, autonomes ou sensoriels: langue commune, sommeil, somnolence dans une pose, arrêts soudains au cours d'une conversation, perte de conscience et autres symptômes.
Caractéristiques des attaques chez les nourrissons
Les premières manifestations de la maladie peuvent survenir à tout âge, mais la plupart des premiers signes d'épilepsie chez les enfants se développent pendant la petite enfance et les années préscolaires. Souvent, les «débuts» de crises convulsives chez les nourrissons sont observés dans le contexte d’une augmentation de la température corporelle, de la peur ou sous l’influence de facteurs externes.
Les manifestations de l'épilepsie chez les nourrissons de nourrissons sont insidieuses et sont dans la plupart des cas masquées par d'autres maladies ou phénomènes physiologiques.
Les premiers symptômes de la maladie chez les nourrissons comprennent:
- jambes et poignées non rythmiques à auto-contraction;
- contractions rythmiques prononcées, petites et rapides des muscles d'une moitié du visage, passant à la jambe et au bras du même côté;
- arrêt soudain du regard du bébé («lag») à court terme ou arrêt soudain de tout mouvement de l’enfant (repli sur lui-même);
- en tournant la tête et les yeux sur le côté, ce qui s'accompagne souvent d'un abduction unilatérale du bras dans la direction du virage;
Comment se manifestent les différents types et formes d'épilepsie chez les enfants d'âge préscolaire et scolaire
À ce jour, les experts ont identifié plus de 40 formes d'épilepsie, qui diffèrent par les symptômes cliniques, l'âge d'apparition des premiers signes de la maladie et l'évolution de la maladie: formes d'épilepsie bénignes ou pronostiquement défavorables chez l'enfant.
Le diagnostic opportun - la définition correcte de la forme de la maladie par un épileptologue spécialisé - revêt une importance particulière. La stratégie thérapeutique et le pronostic de l'évolution de la maladie en dépendent.
Les symptômes cliniques de l'épilepsie chez les enfants dépendent du type de crises et de la forme de la maladie.
Il existe deux formes principales d'épilepsie: "grande" et "petite" - la classification est basée sur la nature des attaques.
Épilepsie vraie (idiopathique ou «grande») chez l'enfant
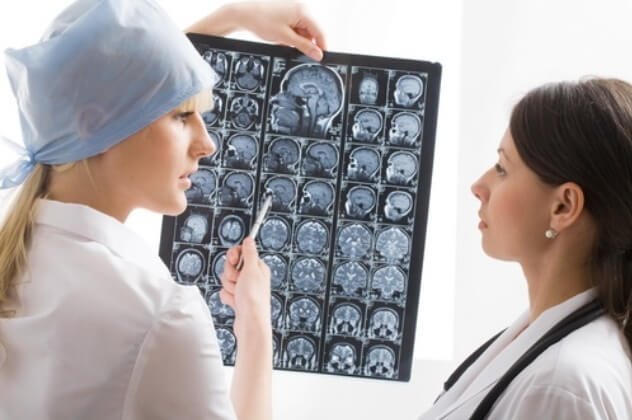
Cette maladie est caractérisée par des convulsions généralisées sous forme de convulsions toniques (redressement et immobilité de groupes musculaires individuels), de convulsions cloniques (contractions musculaires de divers groupes musculaires) ou par le passage d'un type de convulsions à un autre (convulsions cloniques-toniques). Le plus souvent, une "grosse" attaque est accompagnée d'une perte de conscience, d'un arrêt respiratoire, de bave, d'une miction involontaire. Parfois, une crise généralisée s'accompagne d'une morsure de la langue avec libération de mousse sanguinolente de la bouche et d'une perte de mémoire après la crise.
Absannaya ou "petit"
Absansa est un type d'attaque d'épilepsie. Cette pathologie se poursuit par des crises locales (focales ou partielles) dans lesquelles un certain groupe de muscles est impliqué dans le processus. En règle générale, elles se caractérisent par une «décoloration» de l’enfant dans une posture, la tête tournée dans un sens avec arrêt du regard, parfois caractérisée par des contractions d’un groupe musculaire leur atonie nette (relaxation). Après la fin de l'attaque - l'enfant ne ressent pas de décalage dans le temps et continue les mouvements ou la conversation commencés jusqu'à la crise, sans s'en souvenir.
Les absences chez les enfants peuvent aussi se manifester sous la forme:
- sensations auditives, gustatives ou visuelles inhabituelles;
- épisodes de maux de tête spastiques ou de douleurs abdominales, accompagnés de nausée, transpiration, augmentation de la fréquence cardiaque ou de la fièvre;
- troubles mentaux.
Épilepsie nocturne (frontale)
Selon l'heure d'apparition d'une attaque, il y a:
- l'épilepsie de veille;
- l'épilepsie nocturne chez les enfants, dont les symptômes ne se manifestent que pendant le sommeil;
- l'épilepsie avant le réveil.

La nuit est considérée comme la forme la plus bénigne de la maladie et se traite facilement. Les attaques en rêve indiquent clairement la localisation du foyer épileptique dans les lobes frontaux du cerveau (épilepsie frontale).
Avec le développement de la forme nocturne de la maladie, il est important de poser en temps voulu le bon diagnostic. Il est donc nécessaire de savoir reconnaître l’épilepsie chez un enfant, de consulter un spécialiste et de prescrire un traitement à long terme.
Les crises d'épilepsie nocturnes se manifestent par:
- les parasomnies, qui sont les jambes qui tremblent quand on s'endort, qui surviennent involontairement et sont souvent associées à une déficience de mouvement à court terme au réveil;
- dire et somnambulisme, qui sont souvent accompagnés d'énurésie nocturne et de cauchemars. Ces symptômes sont caractéristiques des enfants et guérissent avec l'âge. Lorsque ces symptômes persistent à l'âge adulte, la forme de la maladie devient plus grave et se manifeste par une agressivité au réveil ou des lésions corporelles. Les patients au réveil ne se souviennent de rien.
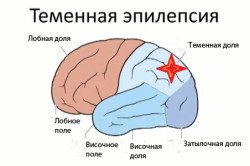
Rolandic
L'épilepsie rolandique est considérée comme la forme la plus commune, bénigne et héréditaire de la maladie.
Les symptômes de la maladie se manifestent dans l'enfance ou adolescence de 2 à 14 ans (généralement de 4 à 10 ans). L'apparition de signes est associée à l'apparition d'un centre d'excitabilité accrue dans le cortex de la région temporale centrale du cerveau (Roland sulcus).

Les symptômes de l'épilepsie rolandique chez les enfants se manifestent:
- aura sensorielle (précurseurs d'une attaque) se présentant sous la forme d'un sentiment unilatéral de picotement, de picotement ou d'engourdissement ou de picotement au niveau des gencives, des lèvres, de la langue, du visage ou de la gorge;
- l'épilepsie attaque se manifeste elle-même sous la forme de convulsions sur un côté du visage ou de contractions unilatérales courtes des muscles du larynx et du pharynx, des lèvres et / ou de la langue, qui s'accompagnent d'une salivation accrue ou de troubles de la parole.
La durée d'un épisode d'épilepsie rolandique est en moyenne de deux à trois minutes. Au début du développement de la maladie, les crises épileptiques se produisent plus fréquemment et se reproduisent plusieurs fois par an. Elles apparaissent moins fréquemment (célibataires) avec l’âge et s’arrêtent complètement.
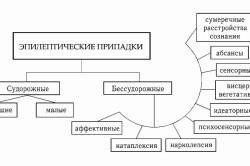
Épilepsie temporale
Ce type d'épilepsie se développe lorsque le foyer épileptique est situé dans les régions temporales du cerveau. Il apparaît à un âge précoce après un traumatisme à la naissance ou une inflammation due à une infection intra-utérine, dans le contexte d'une neuro-infection antérieure (méningite, arachnoïdite ou encéphalite).
L'épilepsie temporale présente des signes caractéristiques et se manifeste par des crises prolongées et une aggravation de la clinique au fil du temps.
Les caractéristiques de cette forme incluent:
- précurseurs de l'attaque (aura) sous forme de douleur abdominale, nausée, crise cardiaque, arythmies, transpiration accrue, difficulté à respirer, difficulté à avaler;
- attaques simples consistant à tourner la tête et les yeux vers le foyer ou des troubles mentaux: éveil, panique, sensation de changements dans le temps, ralentissement ou accélération, troubles de l'humeur, euphorie, dépression, peurs, désorientation dans l'espace et le soi;
- attaques complexes sous la forme de divers mouvements répétitifs (automatisme) - claquer, tapoter, gratter, cligner des yeux, rire, mâcher, répéter des sons individuels, avaler avec des épisodes de désactivation totale de la conscience et le manque de réponse aux stimuli. Avec une évolution compliquée (maligne) de la maladie, des crises convulsives se rejoignent.
Pour diagnostiquer cette maladie en temps utile, il est nécessaire de savoir comment déterminer l'épilepsie chez un enfant: identifier les premiers et principaux signes de la maladie, la fréquence et la durée du début des crises d'épilepsie, et consulter un spécialiste (enfant neurologue, puis l'épileptologue).
Types de crises
http://mama66.ru
Le cerveau est une collection d'un grand nombre de neurones. À la suite du passage des impulsions, il se produit une excitation périodique des cellules nerveuses et la transmission d'informations aux organes. Le but de la transmission d’impulsion est la performance de toute fonction organique. La période entre la transmission des impulsions - le "repos" - est le moment où un neurone n'est pas capable de transmettre une impulsion.
L'épilepsie est un état d'excitation constante dans un groupe de neurones (focus). Les neurones entourant le foyer parviennent temporairement à contenir l'excitation, mais de temps en temps une impulsion électrique se détache du foyer et excite ensuite tous les éléments structurels du cerveau.
Epipristis survient à ce moment précis et se manifeste cliniquement chez les enfants présentant des symptômes d’inconscience, de contraction musculaire, de défécation et de miction involontaires.

L'excitation totale des neurones est lentement remplacée par le processus d'extinction. Les contractions uniques de groupes musculaires individuels sont le résultat de cet "épuisement". Comment se manifeste la prochaine étape de l'épilepsie chez les enfants? Par type de "mode sommeil": l’enfant commence à comprendre ce qui se passe, peut se plaindre de faiblesse musculaire, ainsi que de douleurs dans certains cas. groupes musculairesrésultant de leur travail excessif lors des contractions. Les enfants sont somnolents, lents et ne se souviennent généralement pas de ce qui s'est passé.
En moyenne, quelle que soit la race, l'incidence de l'épilepsie dans le monde entier est d'environ 0,6 à 1% de la population. L'incidence annuelle d'épilepsie enregistrée varie de 20 à 120 nouveaux cas pour 100 000 personnes par an. Les données sur le nombre de convulsions fébriles enregistrées ne sont pas incluses dans les statistiques.
On notera souvent sa compatibilité avec les pathologies suivantes - paralysie cérébrale, troubles métaboliques héréditaires, syndromes chromosomiques. Près d’un tiers des enfants atteints de paralysie cérébrale souffrent d’épilepsie. Les premiers symptômes de l'épilepsie chez les enfants peuvent apparaître dans la période néonatale (le premier mois de la vie). Le risque de convulsions est maximal au cours de la prochaine période de vie - de 1 an à 9 ans. Si vous comptez toutes les convulsions qui surviennent chez une personne pendant toute sa vie, la moitié d'entre elles surviennent à l'âge de 15 ans.
Causes de l'épilepsie. Qu'est-ce qu'ils sont
La prédisposition génétique est un facteur important, mais non déterminant. Pour les formes d'épilepsie symptomatiques, les raisons principales sont les suivantes:
- infections intra-utérines (en particulier, infection à cytomégalovirus (CMV));
- syndromes chromosomiques;
- traumatisme du système nerveux central dû à l'accouchement;
- anomalies fœtales;
- Lésion cérébrale traumatique (TBI);
- néoplasmes cérébraux;
L'épilepsie chez les nourrissons. Quels sont les symptômes de la maladie?
Il est nécessaire de surveiller attentivement le comportement du nouveau-né, car les symptômes manifestés sont atypiques. Les principaux phénomènes: l’absence de réaction aux stimuli extérieurs, l’absence de réactions oculomotrices et de déglutition.
La préparation convulsive dans la petite enfance est due au développement incomplet du cerveau, à des processus d'inhibition inadéquats et à une sorte de métabolisme. L'âge de la petite enfance est caractérisé par des manifestations cliniques de nature limite. Autrement dit, ils peuvent être à la fois les premiers signes du développement de l'épilepsie chez les enfants et ne pas avoir de lien direct avec celle-ci.
Ces conditions limites sont appelées paroxysmes fébriles, apparaissant sur le fond d'une hyperthermie causée par le SRAS ou toute autre infection.
Le diagnostic différentiel de crises simples et complexes, développées à la suite d'une fièvre, est d'une importance fondamentale pour la prévision de l'évolution de la maladie.

Leurs différences sont principalement dans la fréquence d'occurrence. 9 sur 10 de tous les paroxysmes fébriles sont simples. Ils se caractérisent par les caractéristiques suivantes:
- courte durée du cours (pas plus de 15 min);
- les épisodes sont rares;
- paroxysmes généralisés, revêtant une nature tonico-clonique (étirement et tension des extrémités, accompagnés de leurs secousses symétriques, ainsi que de la perte de conscience).
Crises épileptiques inhérentes :
- durée relativement longue (à partir de 15 min);
- répétabilité pendant la journée;
- caractère focal (focal) - conduisant le regard de côté ou vers le haut, tremblant avec un membre ou même une partie d'un membre, regard figé.
Le pronostic est généralement favorable. À l'âge de 5-6 ans, leur disparition indépendante se produit. Chez 4 à 5% des enfants qui ont eu des crises fébriles au cours de leur histoire, leur transformation en épilepsie est également notée.
Cela s'applique principalement aux crises fébriles complexes. Par conséquent, l'attention accrue d'un pédiatre et d'un neurologue aidera à reconnaître l'épilepsie chez les enfants qui, au moins une fois dans leur vie, ont eu un épisode de crises complexes résultant d'un syndrome fébrile.
Des convulsions "accidentelles" sont possibles dans les situations suivantes. :
- intoxication d'origines diverses;
- choc électrique;
- coups de soleil et de chaleur;
- manque de minéraux dans le sang (calcium, magnésium);
- hypoglycémie chez les enfants diabétiques.
Les principaux signes de la différence entre l'épilepsie et les convulsions chez l'enfant :
- répétabilité;
- les facteurs provoquant le développement des crises sont absents.
La rapidité et la précision du diagnostic de l'épilepsie, sa forme spécifique - la tâche du médecin - l'épileptologue. La tactique de traitement et le pronostic de la maladie en dépendent entièrement.
Le rôle des parents dans le diagnostic est inestimable
Très souvent, pour établir un diagnostic correct, le facteur déterminant est une information objective et fiable fournie au médecin par les proches du patient.
Comment déterminer l'épilepsie chez un enfant? Extrêmement important dans le diagnostic est:
- le moment de la première apparition de l'épipristup;
- sa durée (secondes ou minutes);
- la nature (position des yeux et de la tête, mouvement des membres, relaxation ou tension du corps, taille des pupilles et couleur de la peau, que l'enfant soit conscient ou non lors d'une attaque);
- facteurs de provocation possibles (manque de sommeil, stress, lumière gênante, enthousiasme pour les jeux sur ordinateur, mensis, etc.);
- caractéristiques des réactions comportementales avant et après le paroxysme (sommeil, éveil, anxiété, excitabilité, présence d'une "aura", etc.);
- convulsions (avant ou après le réveil, le jour, avant de s'endormir, la nuit).
Les débuts de l'épilepsie rolandique chez les enfants surviennent à l'âge de 2-14 ans, avec un début maximal de 9 à 10 ans. Dans le même temps, les enfants ont une intelligence normale, sans changements pathologiques dans l'AN de l'histoire, mais la famille avait des patients atteints d'épilepsie (génération plus ancienne).
Symptômes: moteurs et somatosensoriels, localisation principalement au visage. Souvent - dysphagie, hypersalivation, engourdissement unilatéral des joues, paresthésies et contractions toniques de la langue. Parfois, le processus implique des membres du même côté.
Les symptômes de l'épilepsie nocturne chez l'enfant sont plus souvent rares et se manifestent dans des postures inhabituelles pendant le sommeil. Lorsque cela se produit, la tension de différentes parties du corps, la bouche est tordue. Au réveil, les enfants ne peuvent rien dire, même s'ils sont conscients.
"Epistatus" se caractérise par une durée de plus de 15 minutes, ainsi que par un manque de conscience dans l'intervalle.
Cette condition est critique, elle met la vie en danger en raison de la possibilité de développement d'un œdème cérébral. Le développement du statut épileptique est une indication directe de l'hospitalisation.
Diagnostic de l'épilepsie chez l'enfant
L'électroencéphalogramme (EEG) est une méthode de diagnostic importante incluse dans l'étude neurologique d'un enfant lors de la première attaque, sans provocation. Son intérêt est d'identifier les risques de récurrence possibles, ainsi que d'identifier les anomalies focales spécifiques. Il est également important d’évaluer l’efficacité du traitement. Ce type de recherche est très informatif lorsqu'il est mené pendant le paroxysme. La méthode est sûre, non invasive et sans douleur.
Récemment, une technique appelée surveillance vidéo EEG, qui consiste en un enregistrement unique d’une attaque et en un enregistrement EEG, a été utilisée. Lorsque des changements pathologiques dans le statut neurologique pour l'examen IRM est nommé. Cette méthode de recherche par rapport à la TDM est plus informative et plus sûre. Pour une IRM de bébés présentant des signes d'épilepsie, ils sont mis dans un sommeil médicamenteux en raison de leur incapacité à rester immobiles pendant la période d'étude.
Premiers secours lors d'une attaque
Règles de conduite et soins d'urgence pour la manifestation de l'épilepsie chez les enfants :
- S'il est précédé d'une aura, il est nécessaire de coucher l'enfant sur le dos ou sur le sol, en s'affranchissant des vêtements contraignants et en déboutonnant son collier.
- Il ne devrait y avoir aucun objet dommageable à proximité. Il devrait également être isolé de l'eau.
- Calme, manque de panique et mouvements chaotiques. C'est la seule façon de contrôler la situation. Enregistrez les heures de début et de fin de l'attaque.
- La tête doit être tournée sur le côté pour éviter que la langue ne retombe et que la salive ne soit aspirée.
- En cas de vomissement, maintenez-le sur le côté.
- Il ne devrait y avoir aucun objet étranger dans la région de la bouche (spatule, cuillère)!
- Vous devez être présent à côté des convulsions.
- Ne donnez pas les médicaments anticonvulsifs prescrits par le médecin par la bouche - l'enfant ne pourra pas les avaler et l'action ne se produira pas après 30 minutes.
- Ne dérangez pas l'enfant, s'il s'endormait après l'attaque, laissez-le dormir.
- Si l'on soupçonne une attaque fébrile, il est nécessaire de mesurer la température corporelle.
- L'administration rectale est préférable. Avantages: sûr, rapide, facile. Pas besoin de compétences spéciales et de conditions stériles.
La dose de diazépam dans les suppositoires est de 0,2 à 0,5 mg pour 1 kg de poids corporel.
Est-il nécessaire de traiter l'épilepsie?
Bien sur. La raison - il est nécessaire de briser le cercle vicieux, lorsque l'attaque précédente donne le "feu vert" à la suivante. Non prescrit dans le traitement à temps conduit au développement d'un retard mental et au développement retardé de l'activité psychomotrice. La principale condition pour la nomination de médicaments à action anticonvulsive: répétée, stéréotypée, provoquant des attaques spontanées.
Principes de prise en charge thérapeutique de l'épilepsie :
- la monothérapie, c'est-à-dire le traitement avec un seul anticonvulsivant;
- le début du traitement - avec une dose minimale, puis passage à une dose thérapeutique suffisante;
- deux médicaments ne peuvent être prescrits qu'avec l'inefficacité de la monothérapie;
- approche individuelle, la conformité de la drogue à la forme de l'épilepsie et le type de crises;
- réception régulière;
- long cours de thérapie (au moins 3 ans);
- le changement de dose ou l'arrêt du médicament ne sont effectués que par le médecin traitant.
Les conséquences des crises d'épilepsie peuvent varier considérablement, principalement en fonction de leur durée et de leur fréquence. Cependant, les paroxysmes à long terme, y compris l'épistatus, peuvent entraîner la mort inévitable des cellules nerveuses.
Les conséquences sociales ont également une connotation négative prononcée. L'enfant est hanté par la peur d'avoir une crise dans un lieu public, la peur du rejet de son état par ses amis et ses camarades de classe. Par conséquent, beaucoup se replient sur eux-mêmes, essayant de mener une vie isolée et fermée. Pour beaucoup, cet état est une tragédie de la vie. Mais les conséquences de convulsions épileptiques soudaines avec perte de conscience sont particulièrement dangereuses. Ils entraînent des ecchymoses, des blessures, des accidents fatals.
Utile sur le traitement de l'épilepsie en Allemagne
Récemment, j'ai écouté une émission sur l'épilepsie chez les enfants en voiture. Oui, c'est effrayant, mais nous ne vivons pas au 19ème siècle et maintenant tout est possible. L'essentiel est d'être attentif à votre enfant et de faire confiance au médecin. Et en aucun cas n'allez pas chez les mamies-guérisseurs, seul un traumatisme psychologique causera et vous perdrez du temps. Merci à l'auteur pour l'article informatif!
Ici, le plus important, c’est que les mères aient si peur des mots épilepsie qu’elles se disputent avec le médecin. Chacun veut prouver que son enfant a tout ce qu'il veut, mais pas l'épilepsie. Et perdre un temps précieux. Plus l’enquête est menée tôt, plus vite on comprend ce qu’elle était. Est-ce l'épilepsie ou les symptômes d'une autre maladie? Il faut faire confiance au médecin.
Oui, c'est une maladie très terrible. Pauvres enfants. Et particulièrement difficile pour les parents de ces enfants. Combien de nerfs en ont besoin pour supporter. Je voudrais leur souhaiter de la patience.
http://moirody.ru
Quels sont les signes d'épilepsie chez les enfants? L'épilepsie est une maladie chronique. Le plus souvent, il est diagnostiqué à un âge précoce. Le symptôme principal de l'épilepsie est des convulsions spécifiques, qui sont épileptiques. La cause des attaques de la maladie sont des désordres dans le cortex cérébral. Selon les statistiques, ce diagnostic est le plus souvent posé chez le nourrisson.

Causes de l'épilepsie chez les enfants
Plusieurs raisons expliquent le développement de la maladie chez l’enfant. Ils sont divisés en certains groupes.
 Dans certains cas, des modifications de la structure du cerveau sont diagnostiquées, souvent la formation de kystes, de tumeurs et d'hémorragies. Le développement de l'épilepsie entraîne souvent des lésions cérébrales. Ces symptômes sont caractéristiques de la forme symptomatique.
Dans certains cas, des modifications de la structure du cerveau sont diagnostiquées, souvent la formation de kystes, de tumeurs et d'hémorragies. Le développement de l'épilepsie entraîne souvent des lésions cérébrales. Ces symptômes sont caractéristiques de la forme symptomatique.
Cependant, les changements ne se produisent pas toujours dans le cerveau, mais un facteur héréditaire intervient. Cette forme s'appelle idiopathique.
La forme cryptogénique est déterminée s’il n’ya pas de cause évidente de la maladie.
Identifier les causes du développement de la maladie vous permet de prescrire le traitement nécessaire. Cependant, il est important de connaître les symptômes et les signes de la maladie.
Symptômes de l'épilepsie chez les enfants
Il convient de noter que les premiers signes d'épilepsie chez les enfants sont significativement différents de ceux observés chez les adultes. Les attaques ne sont pas le seul symptôme de cette maladie.
Comment une crise d'épilepsie survient-elle chez un enfant?
Un symptôme caractéristique de la maladie est une crise convulsive au cours de laquelle les muscles du corps se contractent soudainement.
 Dans le même temps, la respiration s'arrête pendant un court instant. Après un certain temps, il y a des convulsions et des convulsions qui durent d'environ 10 secondes à 20 minutes. Pendant une attaque, la vessie peut se vider. L’attaque s’arrête aussi soudainement au début. Après cela, le patient "tombe" dans un sommeil profond.
Dans le même temps, la respiration s'arrête pendant un court instant. Après un certain temps, il y a des convulsions et des convulsions qui durent d'environ 10 secondes à 20 minutes. Pendant une attaque, la vessie peut se vider. L’attaque s’arrête aussi soudainement au début. Après cela, le patient "tombe" dans un sommeil profond.
Cependant, le bébé a souvent des convulsions sans convulsions. Par conséquent, la maladie est souvent asymptomatique. Pendant une telle attaque, l'enfant gèle pendant un moment. Caractérisé par un regard vide manquant, il y a un tremblement des paupières. Souvent, le bébé jette sa tête en arrière ou ferme ses yeux. Pendant l'attaque, le bébé ne répond pas aux sons. La durée d'une telle attaque ne dépasse pas 20 secondes.
Tout le monde ne sait pas que l'épilepsie chez les enfants peut survenir sous une forme non convulsive. Par conséquent, aucun traitement en temps voulu n'est effectué.
Quels sont les symptômes de l'épilepsie chez les enfants? Parfois, ils peuvent avoir des crises atoniques. Au cours de cette crise, le bébé perd conscience et les muscles du corps sont complètement relâchés. Extérieurement, une telle attaque ressemble à la syncope habituelle. Il faut savoir que cette syncope peut se produire régulièrement. C'est pourquoi vous devez contacter un pédiatre.
Les nourrissons et les nouveau-nés ont souvent des crampes infantiles. Elles se caractérisent par le fait que les mains mènent involontairement vers la poitrine, tout le corps se penche en avant et les pieds des nourrissons se redressent brusquement. Les spasmes chez les enfants se produisent souvent chez les enfants de moins de deux ans et peuvent disparaître complètement après l’âge de cinq ans. Souvent, l'épilepsie se présente sous une forme différente.
Quels sont les autres signes de la maladie chez les enfants?
 Très souvent, les bébés prédisposés à cette maladie se réveillent la nuit après des cauchemars. On leur diagnostique souvent le somnambulisme. Des maux de tête fréquents peuvent survenir sans raison apparente. Un symptôme caractéristique est la nausée ou le vomissement.
Très souvent, les bébés prédisposés à cette maladie se réveillent la nuit après des cauchemars. On leur diagnostique souvent le somnambulisme. Des maux de tête fréquents peuvent survenir sans raison apparente. Un symptôme caractéristique est la nausée ou le vomissement.
Vous devez faire attention si:
- Le bébé a des troubles de la parole.
- Il est conscient, mais ne peut pas parler.
- Fréquence des maux de tête et des troubles de la conscience.
Bien sûr, beaucoup de cauchemars rêvent la nuit et les maux de tête sont communs à tous. Mais si elles deviennent régulières, alors le bébé devrait être montré au neuropathologiste.
Les principaux types d'épilepsie infantile
La maladie a plusieurs types principaux, chacun procédant à sa manière. La principale cause du développement de cette maladie est une violation de l'activité du cerveau. Comme nous le savons, il existe plusieurs parties du cerveau.
Si la localisation de l'épilepsie est déterminée dans la région temporale, elle se caractérise par une perte de conscience à court terme. Il n'y a pas de convulsions. Cette forme se manifeste en violation des fonctions mentales et motrices.
La forme frontale est caractérisée par des symptômes communs de la maladie. Cependant, il peut y avoir des évanouissements, des manifestations convulsives, ainsi que des phénomènes de somnambulisme.
 Les formes pariétale et occipitale de la maladie ont des manifestations différentes, mais sont moins fréquemment diagnostiquées.
Les formes pariétale et occipitale de la maladie ont des manifestations différentes, mais sont moins fréquemment diagnostiquées.
Près de 80% des cas de la maladie, qui sont déterminés chez les enfants, sont les formes temporale et frontale de la maladie. Dans certains cas, une des zones du cerveau peut être affectée. Dans ce cas, la forme focale de la maladie.
La forme focale symptomatique chez les enfants et les adolescents est rarement diagnostiquée. Le plus souvent, il s'agit d'une complication de la maladie, souvent diagnostiquée chez les enfants plus âgés. La tuberculose, la méningite, les attaques de virus sont également à l'origine du développement de formes symptomatiques ou de lésions cérébrales.
Les changements fonctionnels dans les neurones, en particulier leur grande activité, sont caractéristiques de la forme focale idiopathique. Les neurones du cerveau deviennent trop excitables, plusieurs raisons expliquent le développement de cette forme. Parfois, les bébés naissent avec des anomalies et des anomalies cérébrales. Toutes les femmes ne se conforment pas aux recommandations pendant la grossesse. Souvent, les effets secondaires et l'alcool ont de tels effets.
Il existe une autre forme de maladie souvent diagnostiquée chez les enfants de moins de trois ans. La forme rolandique se développe en raison de la mise au point située dans la zone du sillon de Roland. Il est situé dans le cortex du cerveau.
Signes caractéristiques de l'épilepsie rolandique:
- Le bébé développe un engourdissement de la langue, des lèvres et des joues.
- État convulsif des muscles des bras, des jambes et du visage.
- Problèmes d'élocution.
- Pleine conscience.
- Le développement de la salivation.
- Le développement de saisie la nuit.
 La particularité de cette forme de la maladie est caractérisée par le fait qu'elle passe à l'adolescence. L'épilepsie absolue est le plus souvent diagnostiquée chez les filles âgées de 5 à 8 ans. Chez les garçons, ce diagnostic est rarement posé.
La particularité de cette forme de la maladie est caractérisée par le fait qu'elle passe à l'adolescence. L'épilepsie absolue est le plus souvent diagnostiquée chez les filles âgées de 5 à 8 ans. Chez les garçons, ce diagnostic est rarement posé.
Comment diagnostique-t-on l'épilepsie chez les enfants? Les symptômes de l'épilepsie chez les enfants sont très divers et peuvent donc être confondus avec les signes d'autres maladies. Il est particulièrement difficile d'identifier l'épilepsie chez les nourrissons. Par conséquent, pour le diagnostiquer, sont utilisés:
- électroencéphalographie;
- tomographie par ordinateur.
Le diagnostic doit être posé par le médecin après l'examen. Si les parents soupçonnent que l'enfant a des problèmes de développement, vous devez consulter un neurologue.
Comment traiter l'épilepsie chez un enfant
La thérapie ne vise pas seulement à se débarrasser des attaques de la maladie. Cela devrait être complet. Les médicaments appartiennent au groupe des anticonvulsivants. Si le traitement est observé, dans 30% des cas, il est possible de récupérer complètement. La posologie des médicaments au stade initial du traitement est faible. Si la maladie est grave, la fréquence des attaques augmente.
Si une tumeur est détectée pendant le diagnostic, il est recommandé de procéder à une intervention chirurgicale.
L'épilepsie pédiatrique est une maladie grave, mais elle peut être prise en charge si le traitement est instauré rapidement et si toutes les prescriptions du médecin traitant sont suivies.
http://nervzdorov.ru
K.Yu. Mukhin, P.A. Temin, E.A. Rykov
Luigio Rolando (1773-1831), médecin et anatomiste italien, a donné une description topographique classique du sulcus central du cortex cérébral. Dans l'épilepsie partielle idiopathique de l'enfance, caractérisée par de courtes attaques nocturnes motrices hémifaciales, souvent avec une aura antérieure somatosensorielle et des modifications typiques de l'EEG (adhérences polyphasiques avec localisation dans les zones centrale et temporale moyenne), la zone épileptogène est la partie inférieure du sillon rolandique. L'étude de ce type d'épilepsie partielle idiopathique a débuté il y a plus de 40 ans. Initialement, les modifications spécifiques EEG dans un groupe spécifique d'enfants ont été décrites. En 1952, H. Gastaut a constaté pour la première fois des modifications particulières de l'EEG épileptique chez certains enfants: adhérences polyphasiques avec localisation stricte dans la région périrolandique. La première description clinique des symptômes de l'épilepsie avec adhérences rolandiques (1958) appartient à R. Nayras et M. Weausart. Plus tard, E. Gibbs et F. Gibbs, C. Lombroso, P. Loisea et al. a mené une analyse clinique des symptômes de l'épilepsie avec adhérences rolandiques et a suggéré que cette forme d'épilepsie soit appelée rolandique. Les auteurs ont noté un pronostic favorable et l'absence de symptômes de dommages organiques au système nerveux central.
Fréquence
L'épilepsie rolandique (ER) est l'une des formes les plus fréquentes d'épilepsie infantile. Sa prévalence est de 21 pour 100 000 enfants en bonne santé. La fréquence de réapparition de toutes les formes d'épilepsie avec des débuts allant jusqu'à 13 ans varie selon les sources, de 11,5% à 25%. Sur les 360 patients atteints d'épilepsie observés par R. Lerman, 14,4% ont reçu un diagnostic d'épilepsie rolandique. À titre de comparaison, nous notons que l'une des formes d'épilepsie les plus fréquentes chez l'enfant, la pycnolepsie, n'a été diagnostiquée que chez 10,5% des patients. Il est possible que l'incidence réelle de l'épilepsie rolandique dans la population soit beaucoup plus élevée, car de nombreux patients ont des crises convulsives nocturnes isolées, souvent sans l'attention des parents et des médecins.
La plupart des auteurs notent la prédominance des garçons parmi les patients atteints d'épilepsie rolandique. La proportion de garçons et de filles est en moyenne de 6: 4. Parmi les 100 salles de bal observées par R. Lerman, il y avait 62 garçons et 38 filles.
Clinique
Les débuts de l'épilepsie rolandique varient entre 2 et 14 ans. Dans 83% des cas, RE fait ses débuts à l'âge de 4-10 ans. Dans la très grande majorité des cas, les crises commencent entre la cinquième et la dixième année de vie et atteignent leur maximum à l'âge de neuf ans. L'âge moyen d'attaque est de 9,9 ans. Le début de la maladie avant l'âge de 2 ans n'est noté que dans 8% des cas. Il existe des descriptions uniques du début des urgences au cours de la première année de vie, mais on discute de l’affectation de ces cas à l’ER. Le début de la maladie après 11 ans est également rare, et après 14 ans, de tels cas ne sont pas observés.
La symptomatologie clinique des crises d'épilepsie rolandique est généralement typique. Observations partielles simples (motrices, sensorielles, autonomes), partielles complexes (motrices) et généralisées à nouveau. Les paroxysmes moteurs partiels et / ou sensoriels simples sont les plus typiques.
Les crises partielles simples constituent le "noyau" de l'épilepsie rolandique et sont observées chez 70 à 80% des patients. Le début le plus typique d'une attaque d'aura somatosensorielle: sensation de picotement, d'engourdissement, de «passage d'un courant électrique» est unilatéral dans la région du pharynx, de la langue et du chewing-gum. Suivant l'aura, une crise partielle se développe. Les options suivantes sont possibles: attaques hémifaciales; crampes musculaires faciales toniques, cloniques ou tonico-cloniques unilatérales; attaques pharyngoliques; spasmes unilatéraux de la lèvre, de la langue, du pharynx, du larynx, souvent associés à une anarthrie et à une hypersalivation. Des crises hémifaciales se produisent chez 37% des patients, pharyngooral - dans 53% des cas, le développement d'un statut épileptique aux urgences est rapporté. Souvent, aux urgences, environ 11%. Une description détaillée du statut épileptique chez les patients atteints de ER est présentée en N. Fejerman et A.Di Blasi. Les auteurs ont observé 2 garçons âgés de 8 ans avec un nouveau début à l'âge de 3 ans et qui présentaient depuis longtemps des crises convulsives hémifaciales. La conscience a été préservée, cependant, l'anarthrie et la bave prononcée ont été notées. L'administration orale d'anticonvulsivants à forte dose, ainsi que l'administration parentérale de diazépam ou de lorazépam, n'a eu aucun effet sur le statut épileptique. Seule la prescription de médicaments hormonaux (dexaméthasone) a complètement soulagé les convulsions, mais il est douteux d’attribuer ces cas à l’épilepsie rolandique. Des abcès typiques ont été observés chez l’un de ces patients, chez l’autre lors de marches jacksoniennes; les deux avaient des crises résistantes aux anticonvulsivants. Peut-être que ces cas représentent une forme atypique de ER. E.Roulet et al. a décrit le statut épileptique chez un garçon de 6 ans atteint d'épilepsie rolandique. Sa seule manifestation consistait à baver de façon prolongée en association avec une dyspraxie orolingo-motrice. Des complexes spike-wave, y compris des complexes rolandiques, ont été enregistrés sur l'EEG pendant la salivation. Avec l'augmentation des doses de carbamazépine, la bave s'est arrêtée. Selon les auteurs, le seul symptôme du statut épileptique serait une salivation permanente à long terme chez les patients atteints d'épilepsie rolandique.
Les paroxysmes des urgences sont associés au rythme veille-sommeil. Les attaques nocturnes les plus typiques, apparaissant principalement lorsque vous vous endormez et lorsque vous vous réveillez. Pendant les crises nocturnes, des paroxysmes surviennent pendant la période de réveil (35%), moins souvent - tout autour de la nuit (25%) et pendant le sommeil (20%). Chez 25 à 30% des patients, des crises convulsives sont observées à la fois pendant et après le sommeil. Ce n'est que chez 5 à 25% des patients qu'ils surviennent exclusivement à partir du moment de la veille. Il existe certaines différences dans le type d’attaques en fonction du rythme sommeil-veille. Les crises diurnes sont presque toujours simples, souvent hémifaciales, courtes, nocturnes, généralement plus graves, prolongées, généralement unilatérales ou généralisées. L'évaluation de la conscience chez les patients atteints d'attaques nocturnes est difficile; Le phénomène de l’anarthrie simule souvent une clinique d’arrêt mental. Les crises partielles compliquées d'épilepsie rolandique sont rares et ne représentent qu'environ 5% des cas. Il est possible que, chez certains patients, une «fluctuation» du niveau de trouble avec la connaissance se produise au cours d'une attaque.
J.Aicatdi et J. Shevrie se sont vu attribuer un groupe de patients atteints d'un syndrome épileptique concomitant, dont les symptômes cliniques étaient similaires à ceux de l'épilepsie rolandique. Les patients présentaient des crises hémifaciales et hémicloniques partielles simples, mais en combinaison avec des crises myoniques-astatiques, atoniques et, dans certains cas, des absences. Ce groupe d'observations est classé comme une version typique de l'OM. La fréquence, selon T. Deona et al. est de 5% chez tous les patients atteints d’ER. La position taxonomique de ce syndrome est controversée. Selon J. Aicatdi et J. Chevrie, cette forme d'épilepsie est un syndrome nosologiquement indépendant caractérisé par une clinique clairement définie, des modifications de l'EEG et un pronostic. D'autres auteurs suggèrent la présence d'une variante atypique au sein du service des urgences. Une description détaillée de ce syndrome est présentée. T.Deonna et al. sur l'exemple de 6 patients. Parmi les patients examinés, les garçons ont prévalu. Les attaques ont débuté dans la tranche d'âge de 2 à 7 ans chez des enfants neurologiquement normaux ayant une intelligence intacte. Symptômes cliniques inclus différents types les saisies. Les caractéristiques étaient des crises partielles nocturnes simples, similaires à celles des crises ER, myocloniques-astatiques et atopiques. Un certain nombre de patients avaient des absences atypiques. Les attaques dans tous les cas étaient fréquentes, quotidiennes et ont entraîné plusieurs chutes de patients et des blessures graves. Sur l'EEG, les adhérences rolandiques typiques ont été combinées à des complexes à ondes de pic lentes caractéristiques du syndrome de Lennox-Gastaut. Il y avait une augmentation de l'activité épileptique dans la phase de sommeil lent.
Les symptômes cliniques de l'épilepsie rolandique atypique a propose une variété de syndromes épileptiques: ER (simples pics typiques sochetaniis crises nocturnes rolandiques partielles), le syndrome de Lennox - Gastaut (crises de atones astatique fréquentes, crises d'absence atypiques, combinée avec des complexes lents pic-ondes) et smioklonicheski l'épilepsie crises convulsives, décrites par N.Doose. Cependant, une différence fondamentale entre l'épilepsie rolandique atypique et le syndrome de Lennox-Gastaut et l'épilepsie avec crises myocloniques-astatiques réside dans l'absence de troubles mentaux chez les patients et un bon pronostic. R. Loisea et al. ont mené une étude de suivi chez 13 patients atteints de ER atypique pendant au moins 9 ans. Après une période différente, une rémission complète était obtenue dans tous les cas et la majorité des patients (10) n’avaient pas suivi de traitement antiépileptique régulier. Les auteurs n'ont noté aucun retard dans le développement mental, ni aucun trouble grave du comportement, à la fois pendant la période la plus active de la maladie et pendant la période de rémission, ce qui rapproche ces cas d'un OM typique. Selon les auteurs, avec ce syndrome, un traitement antiépileptique «agressif» devrait être évité en raison d’un bon pronostic.
O.Dulac et al. analysé les cas d’épilepsie partielle avec débuts précoces - entre 8 jours et 3 ans de vie. Parmi les 442 patients, une épilepsie partielle 17c a été détectée avec débuts précoces.Les Prisupy ont été caractérisés comme partiels simples avec une généralisation secondaire fréquente. Selon les auteurs, ces cas sont une forme de transition entre l'épilepsie bénigne du nouveau-né et l'ER. Contrairement aux deux syndromes indiqués, le pronostic était défavorable, les crises ne répondant pas au traitement médicamenteux. Des études ultérieures montreront si les cas d'épilepsie partielle débutant au cours des trois premières années de la vie sont un syndrome nosologiquement indépendant ou un autre "variant atypique" de l'ER.
État neurologique
L'état neurologique des enfants souffrant d'épilepsie rolandique a été suffisamment étudié. L'épilepsie rolandique se réfère à des formes idiopathiques d'épilepsie, dans lesquelles il n'y a aucun signe de lésion organique du cerveau, comme le confirment la plupart des publications. De plus, d'après A. Weaumanoir et al. la présence de tels symptômes est contraire aux critères de diagnostic de l'épilepsie rolandique. Cependant, ces dernières années, grâce à l’introduction de pratique clinique méthodes de recherche neuroradiologiques très efficaces (tomographie par ordinateur, résonance magnétique nucléaire, tomographie par émission de positrons), des rapports isolés ont été rapportés sur la détection de modifications structurelles du cerveau chez des patients atteints d’ER.
Sur les 100 patients observés par P.Lerman et S.Kivity, 4 ont présenté les symptômes d'une lésion organique du système nerveux central: paralysie cérébrale (formes hémiparétique et tétraplégique), microcéphalie, retard mental modéré. Dans tous ces cas, le diagnostic de ER n'était pas douteux et a été confirmé cliniquement et neurophysiologiquement. La pneumoencéphalographie n'a révélé aucune anomalie chez ces patients. S.Blom et J.Heijbet dans 3 (7,5%) des 40 cas d'ER retrouvés chez des patients atteints d'hémiparésie centrale. E.Roulet et al. manifestations notées de dyspraxie orolingomotrice chez un patient atteint d'ER. Des symptômes tels que microcéphalie, strabisme, insuffisance cérébelleuse, retard du développement mental et de la parole, ont été signalés chez un certain nombre de patients atteints d'ER R. Santanelli et autres. Un syndrome d'hyperactivité a été observé chez 16,7% des patients atteints d'ER.
La génétique
R.Warau et W.Wiser furent les premiers à déclarer la présence de cas familiaux d'épilepsie rolandique et suggérèrent un modèle de transmission autosomique dominante avec pénétrance incomplète et dépendance à l'âge. Au cours des dernières décennies, de nombreuses études cliniques et généalogiques ont été menées en cas d'épilepsie rolandique, confirmant la validité de ce concept. Selon les données de la littérature généralisée, 8,9% à 68% des patients avec ER ont des parents atteints d'épilepsie ou des antécédents de crises convulsives, et jusqu'à 30% ont des parents qui ont des pics rolandiques sur l'EEG en l'absence de crises. La fréquence de détection de pics chez les enfants de parents et enfants dont les parents ont souffert est de 25 à 36% (dans la population en bonne santé, 1,4 à 5% [W7]). Cependant, seule une petite partie d'entre eux (12% dans l'étude de R. Brau et W.Wiser) ont été marqués par des accès: les autres étaient cliniquement sains. La nature des crises chez les proches des patients atteints d'épilepsie rolandique peut être différente. Les crises typiques caractéristiques des urgences sont constatées chez 13% des membres de la famille, l'épilepsie d'absence chez 10%, les crises convulsives GSR chez 3% et l'épilepsie focale chez 1%.
Un certain nombre d'études ont étudié la concordance de jumeaux monozygotes le long de l'OM. La concordance la plus élevée a été trouvée dans la présence d'épis rolandiques sur l'EEG, mais pas dans le développement de l'ER elle-même. T. Kajitani et al. ont étudié 3 paires de jumeaux monozygotes âgés de 5 à 11 ans dont l'un des frères et sœurs souffrait de re. Dans tous les cas, une activité épileptique rolandique typique sur l'EEG a été retrouvée, mais aucun des frères et sœurs en bonne santé n'a présenté de manifestation de ER.
Ces dernières années, l'hypothèse d'un héritage multifactoriel de l'épilepsie rolandique a été avancée. Il est basé sur le fait que seulement une petite partie des enfants qui ont des adhérences rolandiques sur l'EEG développent davantage les ER. On suppose que les pics rolandiques et le développement de l'ER sont déterminés par deux gènes différents mais liés. En 1982, O.Eeg-Olofsson et al. ont constaté que les parents, les frères et sœurs et les autres membres de la famille des enfants atteints d’eRe présentaient peu d’antigènes d’histocompatibilité des leucocytes de l’haplotype A1B8. Sa fréquence dans la population générale est extrêmement élevée - Il est possible que cet haplotype joue le rôle de protecteur ou de facteur inhibiteur empêchant le développement des ER.
Recherche intéressante révélant le lien de re avec les convulsions fébriles. E. Franzen et al. a rapporté que chez 20% des enfants souffrant de convulsions fébriles, une activité épileptique focale similaire aux épis rolandiques apparaît à l'avenir sur l'EEG. Aucun de ces patients n'a eu d'autres crises d'épilepsie. Des études sur des jumeaux monozygotes ont également montré un lien génétique possible entre les crises convulsives re et fébriles. M. Lennox-Buchthul a étudié 24 paires de jumeaux monozygotes, dans lesquels moins Un des deux partenaires a eu des convulsions fébriles et a découvert que 20% de leurs frères et sœurs avaient de rares crises nocturnes. À 2 sur 24 on trouve une combinaison d'attaques nocturnes avec des spasmes fébriles. Malheureusement, l'auteur n'a pas identifié ces attaques nocturnes; toutefois, en raison de la nature de leur parcours et de l'âge de leurs débuts, on peut supposer qu'il s'agit de cas de MO. Dans l'étude de T. Kajitani et al. parmi 3 paires de jumeaux monozygotes, 3 avaient 5 et 5 crises fébriles. Ces travaux peuvent indiquer un lien génétique entre ER et crises convulsives fébriles. Cette hypothèse est confirmée par la similitude clinique des deux syndromes donnés: un déterminisme génétique élevé, un début «dur» dépendant de l'âge, un pronostic bénin.
Le rôle des facteurs environnementaux exogènes dans le développement de l'épilepsie rolandique reste flou. H.Doose et W.Baier ont suggéré que les facteurs génétiques dans les urgences déterminent le seuil bas pour la préparation du cerveau à la convulsion. Dans le même temps, les enfants en bonne santé présentant des adhérences rolandiques sur l'EEG étaient considérés comme des porteurs subcliniques à seuil convulsif faible. Selon les auteurs, l'impact de divers facteurs exogènes sur l'ontogenèse, ainsi que la liaison avec d'autres gènes, pourraient contribuer à la manifestation clinique de la maladie chez ces patients et avoir une certaine influence sur son évolution. T. Kajitani et al. décrit 3 frères et sœurs autochtones, chacun d'eux ayant des pointes rolandiques typiques sur l'EEG. À l'un des frères et soeurs, RE a fait ses débuts à l'âge de 4 ans lors d'une maladie fébrile, une autre a eu une seule attaque à l'âge de 10 ans lorsqu'elle a regardé la télévision, la troisième a passé la "période critique" pour ses débuts et était en bonne santé. Cette observation illustre le polymorphisme des manifestations cliniques de la maladie, pouvant dépendre de divers effets exogènes.
Les pathologies de la grossesse et de l'accouchement, les blessures à la tête, la neuroinfection, etc. peuvent être des facteurs qui transforment le portage subclinique en ER. Cependant, plus de la moitié des enfants ayant des envois de fonds n’ont aucune indication de la présence de ces facteurs dans l’histoire.
Électroencéphalographie
L’électroencéphalographie est une étude nécessaire à l’objectivation du diagnostic du RE: dans l’EEG de la période intermédiaire, lorsque des complexes caractéristiques typiques «rolandiques» ou «centrifuges» sont détectés avec une activité de base nécessairement préservée. Ces complexes sont des pics diffus de forte amplitude lents ou diffus (100 à 300 mV), souvent suivis d’ondes lentes, d’une durée totale d’environ 30 ms (voir figure). Ils ont tendance à se produire en groupes. Ces complexes ressemblent aux dents du QRST ECG. Les complexes rolandiques sont localisés «rigidement»: les régions centrale et centrale. Les complexes rolandiques peuvent être observés sous forme unilatérale (généralement des crises controlatérales à hémifaciales) - 60% des patients et bilatéralement - 40%. Les complexes épileptiques sont généralement indépendants les uns des autres, mais ils sont rarement observés bilatéralement avec une distribution synchrone avec une amplitude prédominante. les parties.
D.Gregory et R.Wong ont mené une étude sur la localisation exacte des pics rolandiques en utilisant la méthode de cartographie topographique informatisée. Les auteurs ont constaté que le maximum de la "positivité" du dipôle électrique avec l'EM est dans la région centre-temporale, et le maximum de la "négativité" dans la région frontale. Il a été suggéré que des motifs spécifiques d'EEG dans les ER émanent de la zone située dans les parties inférieures du sulcus rolandique, à la frontière avec le sulcus sylvien. Une caractéristique typique des modifications de l'EEG dans les ER est l'instabilité des modèles, leur variabilité d'un enregistrement à l'autre. Les pointes Rolandic peuvent disparaître puis réapparaître, modifier l'harmonie, la configuration, même avec deux enregistrements EEG ultérieurs après une courte période. L'instabilité des profils EEG dans le RE montre, selon J. Aicardi, l'absence de changements organiques dans le cerveau dans le RE. À cet égard, l’absence de pics rolandiques dans une seule étude électroencéphalographique ne peut être considérée comme un argument convaincant pour exclure le diagnostic d’ER en présence d’un type typique de crises. Il est extrêmement important de confirmer que le diagnostic de l'ER est l'étude de l'EEG pendant le sommeil. Environ 30% des enfants souffrant de re, ont des adhérences rolandiques seulement pendant le sommeil. Pendant la somnolence et à tous les stades du sommeil, les complexes rolandiques unilatéraux ont tendance à se transformer en complexes bilatéraux.
Une découverte intéressante pour ER est la détection sur l'EEG avec les complexes rolandiques typiques et d'autres schémas épileptiques. De 10 à 20% des enfants avec ER ont des complexes d’ondes de crête EEG dans d’autres régions du cortex, principalement dans la région occipitale. La morphologie des "complexes occipitaux" est proche du rolandique et de ceux observés dans l'épilepsie focale bénigne avec paroxysmes occipitaux. Selon J. Aicardi, la fréquence des "complexes occipitaux" à Reo est fraternellement proportionnelle à l'âge de l'enfant et n'est pas rare chez les patients de moins de 3 ans. De 7% à 20% des patients avec ER montrent sur l'EEG une activité généralisée typique de l'onde de pic avec une fréquence de 3-4 Hz, se produisant souvent pendant l'hyperventilation. Chez certains patients, l’apparition de complexes à ondes de pic lentes est possible. La plupart des auteurs adhèrent à une opinion unique sur l’absence de corrélation entre la fréquence et la gravité des complexes à ondes de crête sur l’EEG et les caractéristiques de l’évolution de la maladie.
L'étude de l'EEG lors d'une attaque est assez difficile en raison de la faible fréquence des attaques en re-EE. B.Dalla-Bernardina et al. décrit au cours de la nuit attaquer l'apparition sur l'EEG d'activité rapide de faible amplitude dans la région centrale-temporale, se transformant en complexes rolandiques avec la distribution à l'hémisphère entier et avec la couverture ultérieure de l'hémisphère entier.
Recherche neuroradiologique
Les informations sur la recherche neuroradiologique chez les patients atteints d'ER sont rares. H. Gastaut a été l’un des premiers à étudier les données de tomographie assistée par ordinateur chez 15 patients atteints d’ER et n’a constaté aucun changement pathologique. Par la suite, au cours de l'examen neuroradiologique, des modifications structurelles du cerveau ont été observées chez des patients atteints d'ER: élargissement de l'espace sylvien, présence d'une cavité d'un septum transparent (2 cas sur 13), ventriculomégalie, kystes arachnoïdiens (3 cas sur 17). R. Santanelli et al. a présenté une description clinique et neuroradiologique détaillée de 3 cas de RE combinés à des modifications structurelles du cerveau. Chez un patient, un lipome du corps calleux a été détecté, dans un autre (dysplasie spondylothoracique) - une genèse du corps calleux avec ventriculomégalie, dans le troisième - une multitude de calcifications cérébrales, principalement périventriculaires (toxoplasmose congénitale). Tous ces patients présentaient un tableau clinique typique de l'ER et des adhérences rolandiques sur l'EEG.
Les symptômes organiques de lésions cérébrales observés lors d'études neuroradiologiques chez des patients atteints de SE sont difficiles à interpréter sans ambiguïté. Il est possible que ces troubles n'aient pas de relation de cause à effet directe avec le ree et qu'ils soient causés par une association de re avec d'autres maladies - paralysie cérébrale, microcéphalie, toxoplasmose, dysplasie ectomésodermique. Comme dans l'ensemble de la population, les patients atteints d'ER peuvent subir des lésions périnatales du système nerveux central et des lésions cranio-cérébrales postnatales. La fréquence des lésions périnatales du système nerveux central chez les patients atteints de ER est de 6% à 13%, la commotion cérébrale de 4 à 5%, ce qui n’excède pas les données statistiques de la population d’enfants en bonne santé. Une question fondamentalement importante et non entièrement résolue est de savoir si ces facteurs exogènes influencent la survenue et l'évolution de l'ER. Selon J. Aicardi, les atteintes cérébrales constatées chez les patients atteints d'ER ne peuvent être la cause de l'épilepsie, mais constituent des découvertes accidentelles. D'autre part, on ne peut ignorer le fait que chez un certain nombre de patients atteints d'ER, des modifications structurelles du cerveau sont localisées dans les parties inférieures du sulcus rolandique, ce qui n'exclut pas complètement la possibilité de l'existence d'une forme d'épilepsie "symptomatique".
Traitement
Ces dernières années, toute une gamme de médicaments antiépileptiques ont été testés dans le traitement des urgences. La plupart des auteurs recommandent l’utilisation de médicaments agissant principalement sur les formes partielles de l’épilepsie: la carbamazépine (finlepsine, tegretol) et la difénine (phénytoïne). Lors du traitement de l'ER, il est nécessaire d'éviter la polythérapie, ainsi que sur les valeurs des anticonvulsivants à fortes doses. L'évolution bénigne de l'ER, l'absence de troubles intellectuels ménagers chez les patients ne permettent pas de recommander une polythérapie anticonvulsive «agressive» pour le traitement de cette forme d'épilepsie en raison de la possibilité de développement de effets secondaires.
Selon P.Lerman, ainsi que V. Dalla-Bermardina et al., L'un des médicaments de choix pour les urgences est la phénytoïne à faible et moyenne dose. En cas de convulsions uniquement la nuit ou pendant la période de réveil, une seule dose de phénytoïne est recommandée la nuit. Dans tous les cas observés par les auteurs, l'utilisation de doses thérapeutiques moyennes de phénytoïne a entraîné une rémission clinique complète des crises. Selon P.Lerman et S.Kivity, l'efficacité de la phénytoïne dans le traitement des urgences est supérieure à celle de la carbamazépine et du phénobarbital.
La carbamazépine est également très efficace dans le traitement de l'insuffisance rénale et diffère de la difénine par le degré minimal d'effets secondaires du traitement.
Sultiam (ospolot) est efficace et bien toléré par les patients atteints de MO. L'utilisation de barbituriques (phénobarbital, benzonal) est limitée par les effets indésirables fréquents et graves sur la fonction intellectuelle et le comportement des enfants et ne peut donc être recommandée pour le traitement de l'ER.
Ces dernières années, des publications ont montré l'efficacité de préparations à base d'acide valproïque (depakin, konuvuleks) pour le traitement des urgences. L'une de leurs caractéristiques importantes est leur impact positif sur les fonctions intellectuelles et mentales des patients et leur bonne tolérance. Cependant, les études de suivi sur l'efficacité de l'utilisation de valproates pour la MO sont absentes.
La plupart des auteurs estiment que la durée d'utilisation des anticonvulsivants avec ER ne devrait pas dépasser 2 ans depuis la dernière attaque. Durée moyenne traitement de la toxicomanie avec re est de 3,2 ans. Cependant, en cas de convulsions précoces (2 à 3 ans), le traitement doit être poursuivi jusqu'à l'âge de 10 ans, quelle que soit la durée de la rémission. Le traitement est recommandé de nommer seulement après une deuxième attaque. Le maintien sur EEG d'une activité épileptique typique ne peut être la raison de la poursuite du traitement anticonvulsivant, si le patient a une longue rémission.
Prévisions
Une caractéristique particulière de l'ER est un bon pronostic de la maladie. D'après P.Loisea et al. , après 13 ans, les crises de ER ont disparu chez 93,5% des 168 patients examinés et après 16 ans - dans 98,8%. P.Loisea et al. ont étudié en détail l’effet des symptômes cliniques individuels des urgences sur le pronostic de la maladie. Parmi les facteurs susceptibles d’affecter le pronostic, le sexe des patients, l’âge de survenue de l’épilepsie, la fréquence et la nature des crises, leur répartition quotidienne et les caractéristiques de l’état neurologique ont été analysés. Sur les 168 patients atteints d'ER, les antécédents de suivi ont été suivis pendant au moins 4 ans après la dernière attaque. Les facteurs qui influencent la prévision de l'urgence sont mis en évidence: l'âge du début et la fréquence des crises. Il a été constaté que seul le début de l’épilepsie présentait une corrélation statistiquement significative avec les prévisions relatives aux urgences. Il a été établi que la durée de la maladie et la résistance au traitement augmentaient avec l'apparition précoce (jusqu'à 4 ans) et tardive (après 10 ans) de l'épilepsie. Tous les autres facteurs n’affectent pas de manière significative la prévision de la MO.
L'observation prolongée des patients au cours du suivi a révélé la possibilité de crises récurrentes avec ER après une rémission prolongée. Le taux de récidive des crises convulsives à l'âge adulte avec OM est faible - de 1,8% à 4%. La récurrence des crises est plus fréquente chez les femmes lors de l'ajustement hormonal, 3 à 10 ans après le début de la rémission, et n'est pas associée à l'utilisation d'anticonvulsivants. Dans la plupart des cas, il existe des crises convulsives généralisées, uniques ou rares. Une caractéristique importante est l'absence sur l'EEG de pointes rolandiques typiques; d'après P.Loisea et al. EEG chez ces patients est généralement dans la fourchette normale.
Compte tenu du caractère généralisé des crises, de leur apparition longtemps après le début de la rémission, de l'absence d'épis rolandiques sur l'EEG, P. Lerman a suggéré que ces cas n'avaient aucune pertinence pour les ER et constituaient une épilepsie de novo. Ce concept contredit le fait que le taux de récidive des urgences (2 à 4%) dépasse largement la fréquence d'épilepsie dans la population (0,8%). Il est possible que l’interaction de facteurs exogènes avec une diminution du seuil de préparation à la convulsion déterminée par des facteurs génétiques joue un rôle dans la survenue de crises épileptiques à l’âge adulte avec ER. L'un des facteurs «permissifs» peut être une violation de l'équilibre hormonal dans le corps.
Autre cause possible La récurrence des crises peut être l’existence de modifications structurelles persistantes dans le cerveau avec localisation dans les parties inférieures des sillons rolandiques ou sylviens. G. Ambrosetto et al. a décrit la récurrence d'attaques typiques de "rolandicheskie" chez un patient âgé de 21 ans. Le patient souffrait de re 10 ans: convulsions nocturnes simples partielles et secondaires généralisées. Avec 13 ans est venu la rémission, avec 16 detoy arrêté de prendre des anticonvulsivants. À 21 ans, sans raison apparente, la patiente a développé exactement les mêmes crises généralisées nocturnes partielles et secondaires, avec une fréquence de 1 à 4 par mois. Lorsque la tomodensitométrie calculée n'a révélé aucune pathologie. Sur l'EEG à 21 ans et dans 30 ans (en dynamique) - Activité rolandique typique du côté droit et focalisée. La particularité de cette observation est que les nouvelles crises du patient se sont répétées 8 ans après le début de la rémission et étaient typiques de l'ER, mais résistantes aux anticonvulsivants. Selon les auteurs, il existe dans ce cas une forte probabilité que le patient présente des modifications structurelles de la zone rolandique, qui peuvent être détectées par un examen neuroradiologique plus approfondi (résonance magnétique nucléaire, tomographie par émission de positons).
D'autres études montreront le rapport des facteurs génétiques et exogènes dans la détermination de la MO et la possibilité de l'existence de formes «symptomatiques» de RE provoquées par des changements structurels localisés dans la zone rolandique. Il est également nécessaire de mener des études de génétique moléculaire afin de rechercher les gènes responsables du développement de l'ER. Cela permettra de déterminer le lien de réalité avec d'autres formes d'épilepsie idiopathique, telles que l'épilepsie occipitale bénigne, la gastro-encéphalie, l'épilepsie d'absence pédiatrique, etc.
Littérature
I. Badalyan L.O., Temin PA, Mukhin K.Yu. Crampes fébriles. Recommandations méthodiques. M. 1987; 24
2. Karlov V.A. L'épilepsie. M. 1990; 219-223.
3. Sarajishvili P.M., Geladze T.Sh. 1977, 148–149.
4. Aicardi J., Chevrie J.-J. Dev Med Child Neurol 1982, 24: 281 - 292.
5. Aicardi J. Epilepsy chez les enfants 1986; 4
6. Ambrosetto G., P. Tenuper, Baruzzi A. J. Neural Neurosurg Psychiat, 1985; 48: 90.
7. J. Bancaund, D. Collomb et Dell M.B. Rev Neurol 1958; 99: 206 - 209.
8. Beaumanoir A., BallisT., Varfis G., Ansai K. Epilepsia, 1974; 15: 301 - 315.
9. Beaussart M., Favou R. Ibid 1978; 19: 337-342.
10. Beydoun A "index.html" Garofalo E.A "index.html" Drury I. Ibid 1992; 33: 1091-1096.
11. Blom S., Heijbel J. Ibid 1982: 23: 629-631.
12. Bourgeois B.F.D. Ibid 1994 35: 2: 18-23.
13. Bray P.P., Wiser W.C. Pédiatrie 1965; 36: 207 - 211.
14. Classification et terminologie de la commission ILAE. Epilepsia 1989; 30: 389-399.
15. Dalla-Bermardina V., Sgro V., Fontana E. Syndromes épileptiques dans la petite enfance, l'enfance et l'adolescence. Paris 1992; 173 - 186.
16. DeonnaTh., ZieglevA-L., Despland P.-A. Neuropédiatrie 1986; 17: 144 - 151.
17. Deonna Th., Zieglev A.-L., Despland P.-A. Guyvan Mellé. L'épilepsie 1986; 27: 241-247.
18. Doose N., Baier W.K. Egor J Pediatr 1989; 149: 152-158.
19. Les syndromes d'épilepsie de Doose H. pendant l'enfance, l'enfance et l'adolescence. Paris 1992; 103-114.
20. Dreifuss F.E. L'épilepsie 1994; 35: 2: 30 - 34.
21. Dulac O., R. Cusmai, Olivera K.de. Ibid 1989; 30: 798 - 801.
22. Eeg-Olofsson O., Safwenbery J., Wigrtz A. Ibid 1982; 23: 27 - 34.
23. Fejerman N., Di Blasi A.M. Ibid 1987; 28: 351 - 355.
24. Frantzen E., Lennox-Buchthal, M., Nygaakt A., Centre d'électro-céphalogr Neurophysiol1968; 24: 197 - 212.
25. Gastaut H. Rev Neurol 1952; 87: 488 - 490.
26. Gastaut H., Gastaut J.L. L'épilepsie 1976; 17: 325 à 326.
27. Gibbs, E.L., Gibbs, F.A. Ibid 1960; 1: 448-453.
28. Gregory D.L., Wong P.K. Ibid 1984, 25: 705 - 711.
29. Heijbel J. Épilepsie pédiatrique. Helsinky 1989.
30. Kajitani, T., Nakaltmra, M., Llcaka, K., Kobuchi, S. Advances in Epileptology, 1980; 4: 171-175.
31. Kajitani T., Kimura T., Sumita M., Kaneco M. Brain Dev 1992; 14: 230 - 234.
32. Lennox-Buchthul M. Epilepsia 1971, 12: 197-221. 33. Lerman P., Kivity S. ArchNeural 1975; 32: 261-264.
34. Lerman P. Syndromes épileptiques dans la petite enfance, l'enfance et l'adolescence. Paris 1992; 173 - 186.
35. Loisea P., Duche V., Cordova S. et al. L'épilepsie 1988; 29: 229 - 235.
36. Lombroso C.T. Arch Neurol 1967; 17: 52 - 59.
37. Luders, N.O., Lesser R.P., Dinner D.S., Morris H.H. Épilepsie - syndromes électrocliniques. Berlin 1987; 303 - 346.
38. Mambelli M., Moscano F., Maroni P. et al. Bulll. Liga int. L'épilepsie 1985; 51/52: 79-80.
39. Morikawa, T., Osawa, T., Ishihara O., Seino M. Brain Dev 1979; 1: 303 - 346.
40. Nayrac P., Beaussart M. Rev Neurol 1958; 99: 201 - 206.
41. Panayiotopoulos S.R. J Neurol Neurosurg Psychiat 1993; 56: 2 - 5.
42. Roulet E., Deonna Th., Despland P.A. Epilepsia 1989; 30. 564 - 568.
43. Santanelli P., Bureau M., Magaudda A. et al. Ibid 1989; 30: 182 - 188.
Partiel bénin l'épilepsie avec des adhérences centrotemporales (épilepsie rolandique) est une forme courante d'épilepsie partielle chez l'enfant avec un pronostic favorable. Les manifestations cliniques, les données EEG (complexes rolandiques) et l'absence de modifications pathologiques au cours de la neuroimagerie sont des signes caractéristiques de l'épilepsie rolandique, ce qui permet de distinguer facilement cette forme d'épilepsie des crises partielles complexes.
Épilepsie rolandique débuts à l’âge de 2-14 ans, le maximum de débuts est fixé à 9-10 ans. La maladie se développe chez les enfants d'intelligence normale, sans aucun signe de maladie du système nerveux dans l'histoire et sans altération de l'état neurologique. Il y a souvent des signes d'épilepsie dans l'histoire familiale. En règle générale, les attaques sont partielles et se caractérisent par des symptômes moteurs et somatosensoriels, généralement localisés dans la région du visage.
Oropharyngé les symptômes comprennent la contraction tonique et les paresthésies de la langue, un engourdissement unilatéral des joues (surtout le long des gencives), des patients émettant des sons étranges, des dysphagies et une salivation excessive. Des contractions tonico-cloniques unilatérales des muscles du bas du visage accompagnent souvent les crises oropharyngées et sont souvent associées à des convulsions cloniques ou à des paresthésies aux extrémités du même côté. La conscience peut être maintenue ou perturbée; généralisation secondaire possible de la crise partielle.
Environ 20% des enfants n'en ont qu'un. une attaque; chez la plupart des patients, les crises d'épilepsie sont rares et chez 25% des enfants atteints d'épilepsie rolandique, des attaques en série fréquentes se produisent. Avec l'épilepsie rolandique, des crises apparaissent pendant le sommeil chez 75% des patients, alors que des crises partielles complexes se produisent pendant la veille.
Changements dans l'EEG à rolandic ont une valeur diagnostique importante et se caractérisent par la répétition de complexes rolandiques (foyers de pointes) localisés dans la région centre-temporale ou rolandique en combinaison avec une activité normale d’enregistrement en arrière-plan. Les anticonvulsivants sont nécessaires chez les patients présentant des crises fréquentes, mais les médicaments antiépileptiques ne doivent pas être prescrits automatiquement après la première attaque.
Drogue de choix sert de carbamazépine; la carbamazépine continue à être reçue pendant au moins 2 ans ou entre 14 et 16 ans, car à cet âge il y a rémission spontanée de l'épilepsie rolandique.
Encéphalite de Rasmussen
Encéphalite de Rasmussen L'encéphalite subaiguë est l'une des causes de l'épilepsie partielle dans le continu. Les premières crises focales antérieures peuvent être associées à une maladie fébrile non spécifique; les crises peuvent survenir fréquemment ou être permanentes. La maladie fait ses débuts, généralement jusqu’à 10 ans. Peut-être le développement de l'hémiplégie, l'hémianopsie et l'aphasie. Sur EEG, l’activité paroxystique diffuse est enregistrée en association avec un ralentissement de l’activité principale de l’enregistrement en arrière-plan.
Encéphalite de Rasmussen - une maladie évolutive, potentiellement mortelle, cependant, une guérison spontanée avec un déficit neurologique sévère est plus caractéristique. La base pathogénique de la maladie peut être la formation d’autoanticorps qui se lient aux récepteurs glutamatergiques et provoquent leur stimulation. Certaines études ont noté la présence de CMV dans le tissu cérébral de patients atteints d’encéphalite de Rasmussen.
L'épilepsie focale bénigne est plus courante en épileptologie pédiatrique. Ils surviennent chez environ 25% des enfants atteints de convulsions apyrétiques. En règle générale, rares, les attaques nocturnes s’arrêtent entre un et trois ans après les débuts. Chez de nombreux patients, la maladie ne se manifeste que dans la vie, avec une attaque courte ou longue, voire un épisode d'épilepsie. Souvent, les manifestations végétatives critiques se produisent - isolément (crises épileptiques autonomes) ou en association avec d'autres symptômes. Des antécédents de crises fébriles ne sont pas rares. L’état et l’intelligence neurologiques pendant l’examen se situent généralement dans les limites de la normale, bien que certains enfants au stade actif de la maladie puissent présenter des troubles cognitifs réversibles. Ces neuroimagerie sans fonctionnalités.
En dépit des crises épileptiques rares, des perturbations assez prononcées sous forme de pointes de forte amplitude sont notées dans l'EEG. L'EEG normal est rarement enregistré, auquel cas il est souhaitable de mener une étude EEG de sommeil. Des modifications similaires de l'EEG liées à l'âge sont également observées chez 2 à 4% des enfants d'âge scolaire cliniquement en bonne santé ou des enfants à qui une étude EEG a été prescrite pour d'autres raisons.
On connaît aujourd'hui les syndromes d'épilepsie focale bénigne chez l'enfant:
- Epilepsie bénigne de l'enfance avec adhérences centrotemporales (crises rolandiques)
- L'épilepsie occipitale idiopathique, un type de Gasto (la forme dite «pure» de l'épilepsie occipitale idiopathique, est plus fréquente chez les enfants plus âgés)
- Syndrome de Panayotopulos (assez fréquent, avec symptômes ictaux prononcés, isolé relativement récemment, de sorte que les attaques sont souvent interprétées à tort comme non épileptiques)
Les caractéristiques cliniques et EEG d'un syndrome peuvent changer avec le temps, évoluer en un autre syndrome, et une combinaison simultanée des symptômes de différents syndromes est possible. La relation étroite entre ces syndromes suggère l'existence d'un syndrome de prédisposition bénigne dépendant de l'âge aux crises épileptiques chez les enfants, qui peut être interprété comme faisant partie d'un spectre biologique plus large, comprenant également les crises fébriles et les crises néonatales bénignes.
Le kit pédagogique sur les épilepsies
Les syndromes épileptiques
Par C. P. Panayiotopoulos
Le kit pédagogique sur les épilepsies a été réalisé grâce à une subvention à l'éducation sans restriction d'UCB Pharma SA.
Nous vous recommandons d’accepter des traitements dans ces volumes.
Initialement publié par MEDICINAE
21 Cave Street, Oxford OX4 1BA
Première publication en 2006 et réimpression en 2007
Revu et révisé en juin 2008 par Steven C. Schachter, MD

Populaire
- Médicaments contre le rhume: quels sont les médicaments les plus efficaces et les plus sûrs?
- Écoulement muqueux blanc après l'accouchement
- Qu'est-ce que le cancer de la bouche, son profil, ses symptômes et son traitement
- Comment traiter l'herpès sur les lèvres et le corps des remèdes populaires
- Détails sur les antibiotiques fluoroquinolones et les noms de médicaments
- Premiers soins pour les entorses et les fractures
- Cheesy blanc décharge des femmes - un signe de muguet
- Variantes des pertes blanches avant la menstruation et toutes les raisons de leur apparition
- Espérance de vie pour le cancer du sein
- Conséquences de l'introduction du vaccin antipoliomyélitique